Research interested and projects
How cellular physiology is controled by basic molecular and cellular pathways regulating the biology of potassium-selective ion channels
Potassium-selective ion channels are key regulators of neuronal and muscle function. The SPARK team is focused on understanding (1) the cellular mechanisms that control the number, the activity and the subcellular localization of potassium-selective ion channels, and (2) how their dysfunction can be linked to diseases, such as cardiac arrhythmias and neurodevelopmental disorders.
To do so, we are combining the genetic tools available in C. elegans (e.g., forward and reverse genetics, CRISPR/Cas9 genome engineering), with biochemistry, light microscopy and electrophysiology.
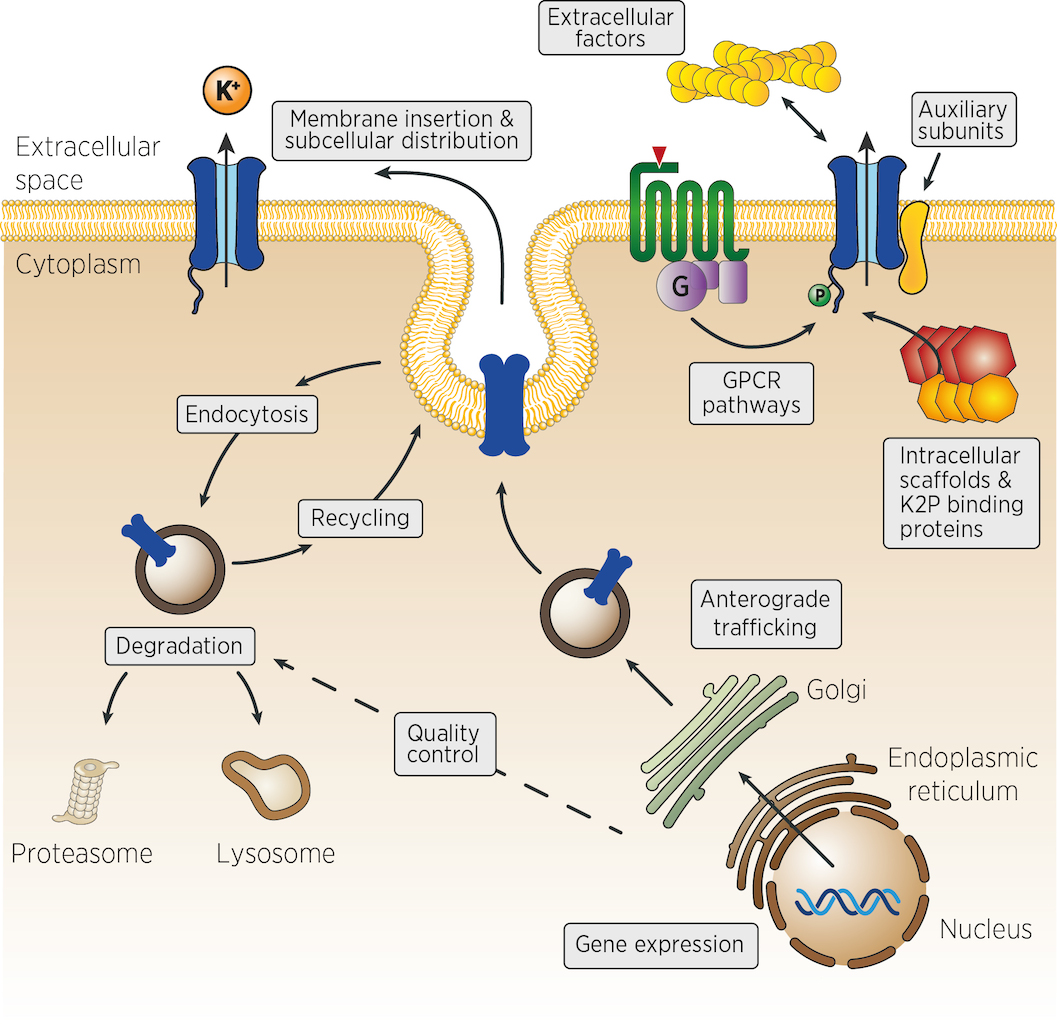 Many different mechanisms could control the number, the activity and the localisation of K+ channels in vivo. From the control of channel expression, biosynthesis and trafficking, to the regulation of membrane insertion, recycling and channel activity at the cell surface (figure). Very few experimental strategies can identify, at once, genes acting in such diverse aspects of cellular physiology. We propose that genetic strategies could address many of these levels of regulation.
Many different mechanisms could control the number, the activity and the localisation of K+ channels in vivo. From the control of channel expression, biosynthesis and trafficking, to the regulation of membrane insertion, recycling and channel activity at the cell surface (figure). Very few experimental strategies can identify, at once, genes acting in such diverse aspects of cellular physiology. We propose that genetic strategies could address many of these levels of regulation.
Genetic screens in model organisms such as flies and worms are ideally suited to dissect complex cellular processes and gene regulatory networks in vivo. C. elegans has proven a powerful, integrated model system for the study of synaptic function, development, aging, cell death or gene regulation by microRNAs and RNA interference. Due to its short life cycle (< 3 days), C. elegans is well suited for large-scale genetic screens. In addition, most mutants of neuronal genes are viable, and even paralyzed worms are able to develop and reproduce.
Recent estimates show that 40% to 75% of human disease genes have homologues in C. elegans. Conversely, about 38 % of the ~21,000 predicted genes of C. elegans have clear homologues in humans (Culetto and Sattelle, 2000; Silverman et al., 2009; Shaye and Greenwald, 2011). Because of this evolutionary conservation, the genes or pathways that regulate K+ function in C. elegans should enable us to identify genes or mechanisms that are also relevant to the regulation of vertebrate potassium channels in excitable and non excitable cells.
Questions that we would like to solve
- What are the cellular mechanisms that control the distribution of K+ channels in vivo in C. elegans?
- Are these pathways conserved in other organisms, including vertebrates?
- How are the number and the activity of K+ channels at the cell surface controlled?
- Are there specific factors that differentially regulate K+ channels in distinct tissues or cell types?
- Do regulators of K+ channels also control the biology of other ion channel families?
Polarity and Membrane Architecture of C. elegans Muscle Cells
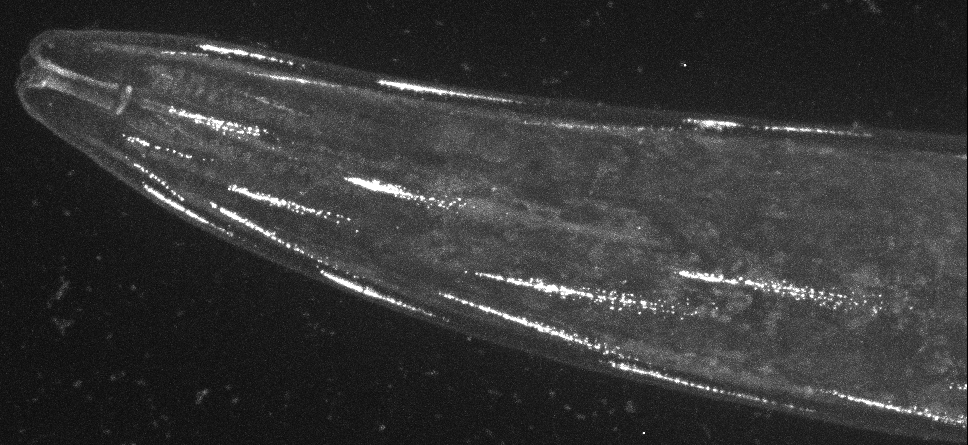
Cell polarity mechanisms create specialized membrane domains with unique protein compositions and functions. For example, most ion channels are addressed to specific compartments within excitable cells. By systematically analysing the localization of over a dozen potassium channels and other membrane proteins, we recently discovered that the plasma membrane of C. elegans muscle cells is polarized (Peysson et al., Nature Communications 2024).
CRISPR-based labeling of the TWK-28 channel revealed and unexpected and fascinating subcellular localisation pattern. TWK-28 channels are found at the anterior tip of individual muscle cells, revealing an entirely unforeseen asymmetry in the organisation of the C. elegans muscle membrane.
Our results reveal, for the first time, the unsuspected complexity of the C. elegans muscle membrane and establish a genetically tractable model system to study cellular polarity and membrane compartmentalization in vivo.
Despite decades of detailed work on the development, structure and function of C. elegans muscle cells, this remarkable case of cellular polarization was completely unsuspected. It is also the first example of tissue-scale planar polarity in this animal. Surprisingly, classical planar polarity proteins are not required. Instead, we show the involvement of a non-canonical Wnt-Ror-Dvl pathway. This conserved but poorly understood pathway has been implicated in morphogenetic processes and is deregulated in various cancers.
In addition, we also discovered that the dystrophin-associated protein complex (DAPC) organizes distinct membrane sub-compartments within the sarcolemma. This observation raised new questions about the way DAPC complexes are formed, about their protein composition and how they can selectively recruit – or “triage” – proteins to different membrane domains.
The key questions we want to address are: (1) What are the downstream effectors of the Wnt-Ror-Dvl signaling pathway that implement muscle polarity? (2) How are distinct membrane domains established and how are their specific protein compositions determined?
Functional Studies of C. elegans Two-Pore domain Potassium (K2P) Channels
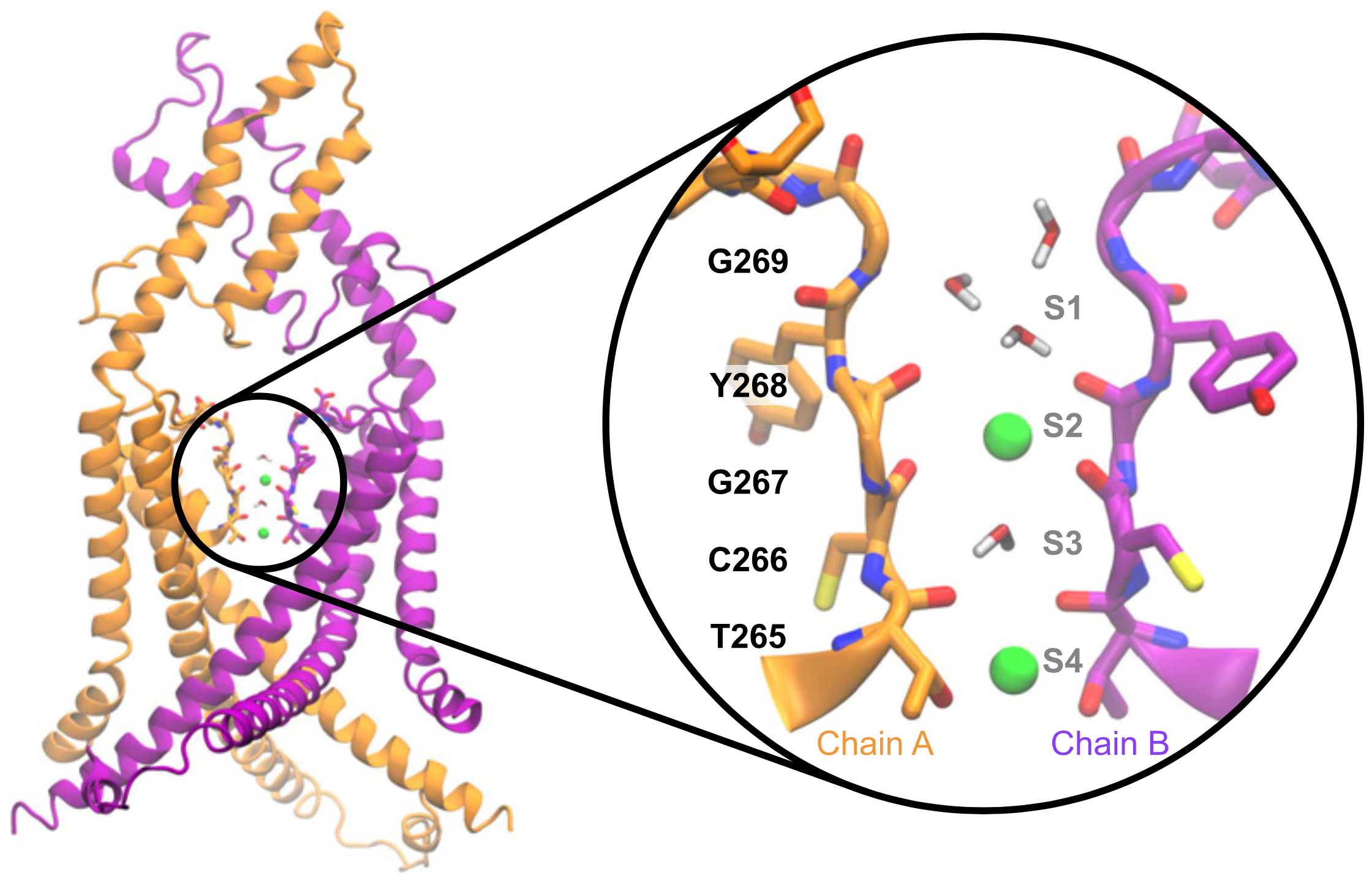
Potassium channels are crucial for regulating cellular excitability, with their unique structure enabling selective ion passage through cell membranes. Gain‐of‐function mutations that modulate the activity of ion channels are essential tools to study their function. We have shown that a single conserved residue controls the activity of K2P potassium channels in vertebrates and invertebrates and can be mutated to progressively increase channel open probability (Ben Soussia et al., Nature Communications 2019). This discovery has been pivotal for our projects, as it enabled us to generate the necessary gain‐of‐function knockin lines to study the contribution of K2P channels to the control of cellular excitability.
Three studies stemming directly from this work have been published. Two collaborative studies describe the role of the TWK-40 channel in controlling locomotor and rhythmic behaviors (Yue et al., PNAS Nexus 2024; Meng et al., Science Advances 2024). In Meng et al., we describe a new conceptual framework for understanding how animals transition smoothly between opposing motor states, such as forward and backward movement. In the third study, we describe the functional characterization of an atypical potassium channel, UNC-58, which is the first example of a K2P channel that is constitutively permeable to sodium ions (Andrini et al., PNAS 2024).
Functional studies of rare disease gene variants in C. elegans
Rare human diseases are only rare in name. Over 300 million patients are affected by approx. 7000 rare diseases. A years-long diagnostic odyssey is typical for most rare disease patients. Therefore, functional validation studies are an essential step in determining the pathogenicity of variants identified in patients with rare genetic diseases. Missense mutations pose a specific challenge, as it remains difficult to predict their impact in most cases, and modelling disease mutations, in vivo, in animal models still represents a gold standard.
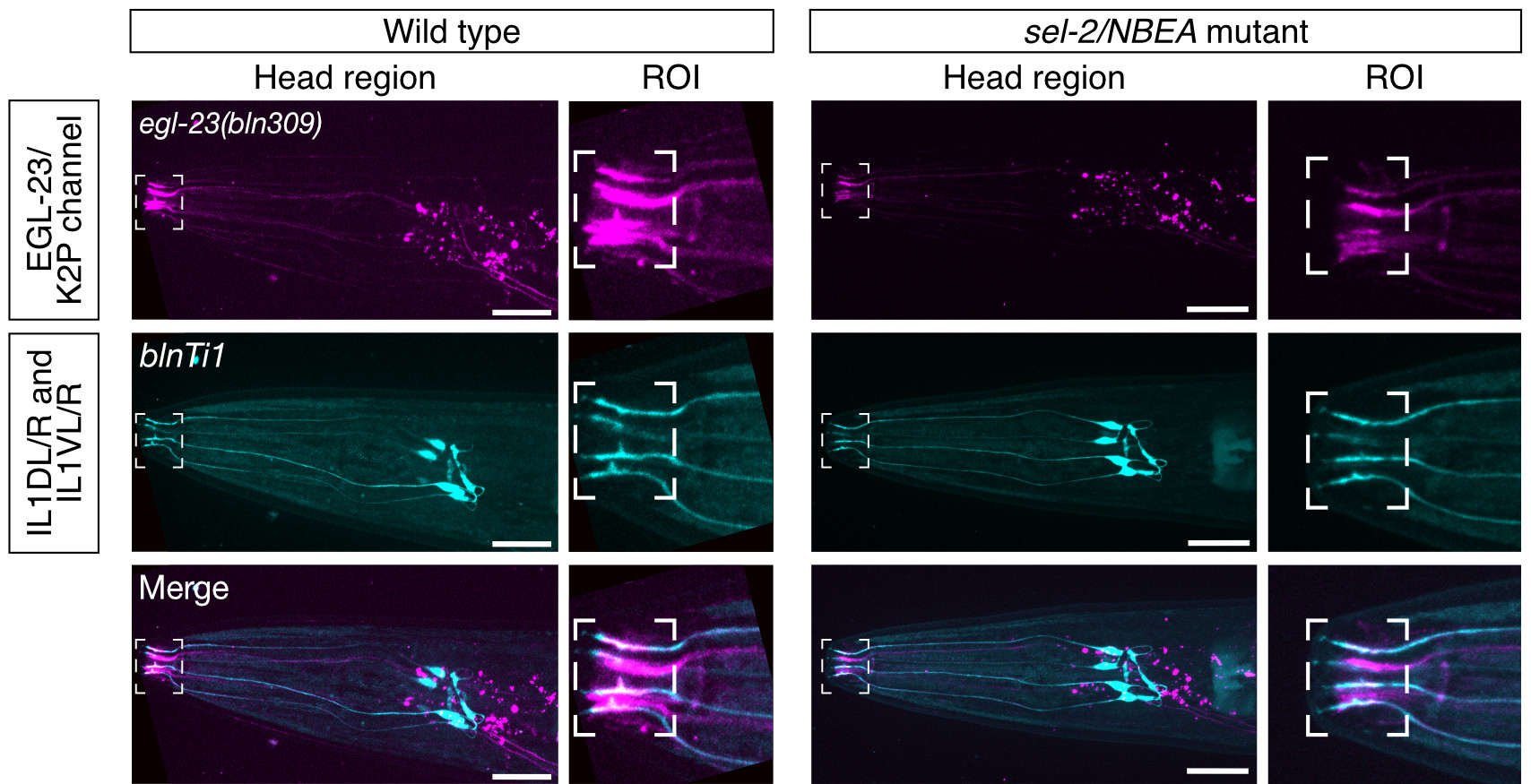
Neurobeachin/NBEA is a 327 kDa brain-specific protein. Dysfunction of this protein is associated with NEDEGE syndrome (Neurodevelopmental Disorder with or without Early-Onset Generalized Epilepsy, OMIM 619157), a very rare neurological disease that was recently identified. Neurobeachin is required for the proper trafficking of different classes of neurotransmitter receptors in vertebrates. We have found it is also required for the biosynthesis of K2P potassium channels in the worm. In collaboration with the team of Tim Schedl (Washington University, St. Louis) and the Undiagnosed Diseases Network (UDN, NIH), we developed the first functional assay for NBEA variants based on these findings. This assay confirmed the pathogenicity of a de novo variant found in a patient under the care of the UDN (Boulin et al., MGM 2021).
Functional analysis of KCNH2 missense variants associated with congenital long-QT syndrome
Congenital long QT syndrome (LQTS) is a hereditary condition responsible for sudden death in young individuals. With a prevalence of approximately 1/2000 births, this syndrome represents the most common hereditary channelopathy worldwide. Sixteen genes have been associated with LQTS. Among these, variants of the potassium channel gene KCNH2 alone account for 30 to 45% of cases. KCNH2 encodes the hERG subunit of the voltage-sensitive potassium channel Kv11.1. This channel is responsible for the primary current during the repolarization phase in cardiomyocytes. Dysfunction of the hERG channel leads to prolonged repolarization and may induce fatal ventricular arrhythmias.
In a study lead by Olga Andrini and Antoine Delinière, we report the clinical and electrophysiological characterization of the novel p.Gly603Ser KCNH2 variant (Delinière et al., Gene 2024). This patient mutation is the first example of a homozygous loss-of-function variant in the pore of the Kv11.1 potassium channel that causes a severe but viable long-QT syndrome with a delayed clinical expression.
This work, conducted in collaboration with the Centre National de Référence des Troubles du Rythme Cardiaque d'Origine Héréditaire de Lyon (CERA, HCL), bridges basic research and clinical insights, facilitating the interpretation of genetic findings for personalized medicine.
Evolutionary Variation in Egg-Laying Behavior of C. elegans
Reproductive behaviors vary considerably among animals—not only between species but also within a single species. However, we still have a limited understanding of the molecular processes that allow neural circuits to evolve within a species to generate such behavioural variation.
We used the egg-laying circuit of C. elegans as a model system to study natural intraspecific microevolutionary variability. In collaboration with Christian Braendle’s team (iBV, Nice), we demonstrated that a mutation in a single gene, the SK potassium channel KCNL-1, results in the loss of environmental sensitivity in several natural isolates. Instead of regulating egg-laying based on the presence or absence of food, these isolates constitutively retain their embryos, which leads to internal hatching. By combining our complementary expertise, we achieved a comprehensive characterization—from the molecular and cellular levels to the evolutionary process – that led to the selection of this paradoxical phenotypic trait (Vigne et al., Science Advances 2021).
Building on this work, we published a second study that characterizes the intraspecific variation of egg-laying behavior in C. elegans (Mignerot et al., eLife 2023). In this study, by analysing a collection of approximately 300 wild isolates, we describe highly variable intra-uterine retention. By applying various pharmacological agents and genetically manipulating endogenous serotonin levels in 10 wild isolates, we demonstrated that this behavioural variation stems from an evolutionarily divergent neuromodulatory architecture.